ご利用にあたってのご注意
このQ&Aに記載の情報は、製品の適正使用にあたっての参考情報であり、すべての患者さん/事例にあてはまるものではありません。
そのため、Q&Aの利用に関して生じた結果については、責任を負いかねますので、ご了承ください。
また、国内で承認されていない効能又は効果/用法及び用量等の情報を含む場合がありますが、弊社としてこれらの使用を推奨するものではありません。
製品のご使用にあたっては、最新の電子化された添付文書をご確認ください。
製品に関してご不明な点がございましたら、弊社ユーシービーケアーズ コンタクトセンター(0120-093-189)にお問い合わせください。なお、本Q&Aを
許可なく複写、複製、転掲、改変、配布等を行うことは固くお断りします。
補体(C5)阻害剤
ジルビスク®
- ジルビスク®皮下注16.6㎎シリンジ
- ジルビスク®皮下注23.0mgシリンジ
- ジルビスク®皮下注32.4mgシリンジ
髄膜炎菌感染症は致命的な経過をたどることがありますので、緊急時に十分に措置できる医療施設及び医師のもとで、あるいは髄膜炎菌感染症の診断及び治療が可能な医療施設との連携下で投与してください。
また、本剤は、全身型重症筋無力症(gMG)に十分な知識を持つ医師のもとで、治療上の有益性が危険性を上回ると判断される場合にのみ投与することができます。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
眼筋型重症筋無力症への使用は、適応外となります。
本剤の効能又は効果は「全身型重症筋無力症(ステロイド剤又はステロイド剤以外の免疫抑制剤が十分に奏効しない場合に限る)」です。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
本剤の投与開始前には、抗AChR抗体が陽性であることを確認してください。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
本剤は補体第5成分(C5)レベルで補体活性を阻害します。
補体カスケードは、抗AChR抗体[免疫グロブリンG(IgG)1及びIgG3 アイソタイプ]により活性化されます。抗AChR抗体陰性患者では補体カスケードが活性化されないため、ジルビスクⓇの効果は期待できません。
また、抗AChR抗体陰性のgMG患者を対象とした臨床試験は実施していません。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
抗アセチルコリン受容体抗体陽性の18歳以上の全身型重症筋無力症患者(MG-ADL総スコアが 6 以上、かつQMG総スコアが12以上)174例(日本人患者16例を含む)を対象に、多施設無作為化二重盲検プラセボ対照試験を実施した。12週間の投与期間を通じて用量を一定とする標準治療(ステロイド、免疫抑制剤)が併用可能とされた。本剤又はプラセボを 1日 1 回皮下投与したとき、主要評価項目である投与12週時のMG-ADL総スコアのベースラインからの変化量は表のとおりであり、プラセボ群に対して統計学的に有意な改善が認められた。また、副次評価項目である投与12週時のQMG総スコアのベースラインからの変化量は表のとおりであった。
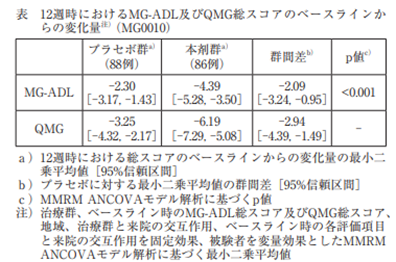
副作用発現頻度は、本剤群で32.6%(28/86例)であった。主な副作用は、注射部位内出血11.6%(10/86例)、注射部位疼痛9.3%( 8 /86例)、注射部位反応、挫傷、リパーゼ増加及び頭痛各3.5%( 3 /86例)であった。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
本剤の投与により、髄膜炎菌感染症を発症しやすくなる可能性があります。髄膜炎菌感染症の既往のある患者へ投与する場合は、患者の状態に十分注意してください。
本剤の投与に際しては髄膜炎菌感染症の初期徴候(発熱、頭痛、項部硬直等)に注意して観察を十分に行い、髄膜炎菌感染症が疑われる場合及び否定できない場合には、直ちに診察し、抗菌薬の投与、本剤の投与中止等の適切な処置を、髄膜炎菌感染症が否定できるまで行ってください。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ適正使用ガイド
電子添文上、特定の背景を有する患者に関する注意の項に、腎機能障害患者の規定はありません。特別な注意喚起は行っておりません。
■腎機能障害患者を対象とした試験(UP0114試験:外国人データ)
重度腎機能障害患者(CLcrが30mL/分未満と定義)及び腎機能正常患者(各8例計16例)を対象に、ジルコプラン0.3mg/kg注)を単回皮下投与したときの薬物動態を評価する第I相、非盲検試験において、本剤投与後、重度腎機能障害患者群及び腎機能正常患者群ともにTEAEの発現は認められませんでした。また、その他の安全性評価項目(臨床検査値、バイタルサイン、12誘導心電図、身体所見)にも臨床的に重要な変動は認められませんでした。重度腎機能障害患者及び腎機能正常患者にジルコプラン0.3mg/kg注)を単回皮下投与したときの安全性が確認されました。
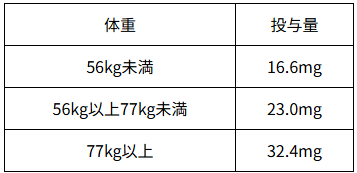
注)ジルビスクⓇで承認されている「用法及び用量」は以下のとおりである。
「通常、成人にはジルコプランとして下表に示す用量を1日1回皮下投与する。」
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
電子添文上、特定の背景を有する患者に関する注意の項に、肝機能障害患者の記載はありません。特別な注意喚起は行っておりません。
■肝機能障害患者を対象とした試験(UP0094試験:外国人データ)
中等度の肝機能障害[Child-Pugh 分類で中等度(7~9点)]患者及び肝機能正常患者(各8例計16例)を対象に、ジルコプラン0.3mg/kg注)を単回皮下投与したときの薬物動態を評価する第I相、単施設、非盲検試験において、全体で3例(18.8%)の患者に5件の治験薬投与後に発現したTEAEが認められ、このうち中等度肝機能障害患者群が2例(25.0%)4件、肝機能正常患者群が1例(12.5%)1件でした。試験期間中に、重篤な有害事象(SAE)、高度のTEAE、試験中止に至ったTEAE及び死亡は認められませんでした。
中等度肝機能障害患者群の1例に副作用として悪心が発現しました。当該事象の重症度は軽度、発現期間は20日間で転帰は消失でした。肝機能正常患者群では、副作用は認められませんでした。また、その他の安全性評価項目(臨床検査値、バイタルサイン、12誘導心電図、身体所見)にも臨床的に重要な変動は認められませんでした。本試験の結果から、新たな安全性の所見及び中等度肝機能障害患者群と肝機能正常患者群間の安全性プロファイルに違いは認められませんでした。
注)ジルビスクⓇで承認されている「用法及び用量」は以下のとおりである。
「通常、成人にはジルコプランとして下表に示す用量を1日1回皮下投与する。」
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
電子添文上、特定の背景を有する患者に関する注意の項に、生殖能を有する者の記載はありません。
なお、gMG患者にジルコプランを投与した場合の妊孕性、生殖能について検討した試験は実施しておりません。
■生殖発生毒性試験
ジルコプランを投与量1、2及び4mg/kg/日で妊娠中のカニクイザルに1日1回皮下投与したとき、母動物、胚・胎児発生期(妊娠20~100日)、並びに出生前及び出生後の発達時期(妊娠20日から出産を経て授乳/出生後90日まで)に毒性は誘発されませんでした。出生児の成長と発達に悪影響はなく、あるいは形態学的所見に毒性学的懸念はありませんでした。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与してください。妊婦に対する安全性は確立されていません。妊婦へのジルビスクⓇ投与に関するデータはありません。
■生殖発生毒性試験
ジルコプランを投与量1、2及び4mg/kg/日で妊娠中のカニクイザルに1日1回皮下投与したとき、母動物、胚・胎児発生期(妊娠20~100日)、並びに出生前及び出生後の発達時期(妊娠20日から出産を経て授乳/出生後90日まで)に毒性は誘発されませんでした。出生児の成長と発達に悪影響はなく、あるいは形態学的所見に毒性学的懸念はありませんでした。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
治療上の有益性及び母乳栄養の有益性を考慮して、授乳の継続又は中止を検討してください。
授乳婦に対する安全性は確立されていません。ジルコプランの母乳への移行、又は乳児が母乳を経口摂取した際の全身への吸収については不明です。
■生殖発生毒性試験
ジルコプランを投与量1、2及び4mg/kg/日で妊娠中のカニクイザルに1日1回皮下投与したとき、母動物、胚・胎児発生期(妊娠20~100日)、並びに出生前及び出生後の発達時期(妊娠20日から出産を経て授乳/出生後90日まで)に毒性は誘発されませんでした。出生児の成長と発達に悪影響はなく、あるいは形態学的所見に毒性学的懸念はありませんでした。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム
電子添文上、特定の背景を有する患者に関する注意の項に、高齢者の規定はありません。
特別な注意喚起は行っておりません。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
本剤は、抗MuSK抗体陽性の患者へは投与できません。適応外となります。
ジルビスクⓇは、抗AChR抗体陽性のgMG(ステロイド剤又はステロイド剤以外の免疫抑制剤が十分に奏効しない場合に限る)の患者に投与可能な薬剤です。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
なぜ、髄膜炎菌ワクチンの接種が必要なのですか。
原則として、髄膜炎菌ワクチンの接種を行わないで本剤を投与することはできません。
原則として、本剤投与開始の少なくとも2週間前までに髄膜炎菌に対するワクチンを接種してください。
なぜ、髄膜炎菌ワクチンの接種が必要なのですか。
本剤は、C5の開裂及びC5bとC6の結合を阻害し、終末補体複合体C5b-9の生成を抑制すると考えられるため、髄膜炎菌をはじめとする莢膜形成細菌による感染症を発症しやすくなる可能性があります。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
本剤の適応は、成人のみです。
小児への適応はありません。小児等を対象とした臨床試験は実施していません。
成人(15歳以上)から投与可能です。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
電子添文には、食前、食後、食間、就寝前といった規定はありません。
本剤は1日1回投与する薬剤であり、可能な限り同一時間帯に投与してください。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
他剤から本剤に切り替える場合に、どのくらいのウォッシュアウト期間が必要かについて検討する臨床試験は実施していません。
また、本剤から他剤に切り替える場合は、本剤の薬物動態、切り替え後の製品の特性等を考慮の上、ご判断ください。
参考までに、ジルビスクⓇ単回投与時の半減期は、183~201時間(約7~8日)です。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
■作用機序
本剤の有効成分であるジルコプランは、C5に結合しC5a及びC5bへの開裂並びにC5b及びC6の結合を阻害することにより、膜侵襲複合体(MAC)の形成及び細胞溶解活性を抑制します。
(詳細)
1. ジルコプランはC5のC5bに相当する部位に高い親和性で結合してC5のC5a及びC5bへの開裂を阻害し、下流の補体活性化を抑制します。
2. ジルコプランはC5のC5bに相当する部位に結合していることから、C5bが形成された場合でもC6との相互作用を立体的にブロックし、MACの形成を第一段階で阻害します。
3. 1、2の2つの作用によりシナプス後膜でのMAC形成が阻害されます。
4. MACの形成と蓄積を阻害することで運動終板への障害が抑制され、神経筋伝達が維持されるものと推測されています。
引用
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ 電子添文
- ジルビスクⓇ皮下注16.6mg/23.0mg/32.4mgシリンジ インタビューフォーム