ご利用にあたってのご注意
このQ&Aに記載の情報は、製品の適正使用にあたっての参考情報であり、すべての患者さん/事例にあてはまるものではありません。
そのため、Q&Aの利用に関して生じた結果については、責任を負いかねますので、ご了承ください。
また、国内で承認されていない効能又は効果/用法及び用量等の情報を含む場合がありますが、弊社としてこれらの使用を推奨するものではありません。
製品のご使用にあたっては、最新の電子化された添付文書をご確認ください。
製品に関してご不明な点がございましたら、弊社ユーシービーケアーズ コンタクトセンター(0120-093-189)にお問い合わせください。なお、本Q&Aを
許可なく複写、複製、転掲、改変、配布等を行うことは固くお断りします。
抗てんかん剤
ブリィビアクト®
- ブリィビアクト®錠25mg
- ブリィビアクト®錠50mg
- ブリィビアクト®静注25mg
本剤のインタビューフォームに以下の記載があります。
I.概要に関する項目
1. 開発の経緯
ブリーバラセタム(以下、BRV)はベルギーのUCB Pharma S.A.でにおいて、レベチラセタム結合部位として同定された脳内のシナプス小胞蛋白質2A(SV2A)への選択的かつ高親和性のリガンドを特定することを目的とした創薬プログラムにて開発された抗てんかん薬である。
2ピロリドン誘導体であるBRVとSV2Aの結合が発作抑制作用に寄与しているものと考えられている。
BRVは最大電撃けいれん発作及びペンチレンテトラゾール誘発けいれん発作などの抗てんかん薬用スクリーニングモデルにおいて発作抑制効果を示し、各種てんかんモデル動物(部分てんかん、薬剤耐性てんかん、全般てんかん及び重積状態)においても、非毒性量で発作抑制作用を示した。
また、動物及びヒトにおいてBRVは100%に近い経口バイオアベイラビリティを示した。以上のことから、欧州及び米国等で臨床試験が開始され、2016年に欧州等で、「成人てんかん患者の部分発作に対する併用療法」として[経口剤50ヵ国、静注用製剤47ヵ国(2025年9月現在)]、また、米国では2017年9月に、「16歳以上の部分発作を有するてんかん患者に対する単剤療法」としても、外挿に基づいて承認を取得した。
本邦において、BRVは海外と同様に、成人てんかん患者における「他の抗てんかん薬で十分な効果が認められないてんかん患者の部分発作(二次性全般化発作を含む)に対する抗てんかん薬との併用療法」に対する適応を取得することを目標として、2008年2月より、ユーシービージャパン株式会社が本邦での開発を開始し、第III相臨床試験(EP0083試験)及びその継続投与試験(EP0085試験)においてBRVの海外臨床試験と同様の有効性及び安全性の結果が確認された。
また、「一時的に経口投与ができないてんかん患者における部分発作(二次性全般化発作を含む)の治療に対するBRV経口製剤の代替療法」を適応とした静注用製剤も開発し、本適応取得のために計画した臨床試験(EP0117及びEP0118試験)を完了した。さらに「てんかん患者の部分発作(二次性全般化発作を含む)に対する単剤療法」の適応についても、併用療法から単剤療法への外挿が認められ、錠剤及び静注用製剤について、併用療法と単剤療法の適応を併せて「てんかん患者の部分発作(二次性全般化発作を含む)」を効能又は効果とする製造販売承認申請を行い、2024年6月24日に承認を取得した。
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
- ブリィビアクト静注25mg インタビューフォーム
本剤のインタビューフォームに以下の記載があります。
I.概要に関する項目
2. 製品の治療学的特性
【経口製剤】
①ブリーバラセタム(以下、BRV)はレベチラセタムと同様に神経終末のシナプス小胞膜に存在する輸送体様の膜蛋白質、シナプス小胞蛋白質 2A(SV2A)に結合して、てんかん発作抑制作用を発揮する。
②各種てんかんモデル動物(部分てんかん、薬剤耐性てんかん、全般てんかん及び重積状態)において、非毒性量で発作抑制作用を示した。
③十分な発作コントロールが得られていない部分発作を有する患者に対し、BRVは抗てんかん薬の使用歴や併用抗てんかん薬の剤数にかかわらず、部分発作のコントロールに有効である。
④ BRVは単剤療法及び併用療法において同じ用法・用量で使用できる。
⑤ 主要な試験において、重篤な有害事象や投与中止に至った有害事象が少なく、性別、年齢、人種等で安全性プロファイルに違いはなかった。
⑥ BRVの薬物動態プロファイルは用量比例性によって特徴付けられるため、治療用量のモニタリングは不要である。
⑦ 抗てんかん薬(以下、AED)及び他の薬剤が血漿中BRV濃度に及ぼす影響はカルバマゼピン(以下、CBZ)フェニトイン(以下、PHT)及び強力なCYP誘導剤であるリファンピシンRFPを除いて確認されず、BRVがAED及び他の薬剤の血漿中濃度に及ぼす影響は、CBZ代謝物及びPHTが上昇したことを除いて確認されなかった。
⑧重大な副作用として攻撃性(0.3%)があらわれることがある。
【静注製剤】
①ブリーバラセタム(以下、BRV)はレベチラセタムと同様に 神経終末のシナプス小胞膜に存在する輸送体様の膜蛋白質、シナプス小胞蛋白質 2A(SV2A)に結合して、てんかん発作抑制作用を発揮する。
②各種てんかんモデル動物(部分てんかん、薬剤耐性てんかん、全般てんかん及び重積状態)において、非毒性量で発作抑制作用を示した。
③十分な発作コントロールが得られていない部分発作を有する患者に対し、BRVは抗てんかん薬の使用歴や併用抗てんかん薬の剤数にかかわらず、部分発作のコントロールに有効である。
④BRV 経口投与を静脈内投与に切り替える、又はその逆の場合、用量の調整をせずに同じ用法・用量の維持が可能である。
⑤BRV 静脈内投与は一時的に経口投与が不可能な患者の治療継続を可能にする。
⑥BRV 経口剤と静注用製剤の安全性プロファイルは同様である。
⑦重大な副作用として攻撃性(0.3%)があらわれることがある。
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
- ブリィビアクト静注25mg インタビューフォーム
本剤の電子化された添付文書に以下の記載があります。
【経口製剤】
4.効能又は効果
てんかん患者の部分発作(二次性全般化発作を含む)
【静注製剤】
4.効能又は効果
一時的に経口投与ができない患者における、下記の治療に対するブリーバラセタム経口製剤の代替療法
てんかん患者の部分発作(二次性全般化発作を含む)
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に以下の記載があります。
【経口製剤】
6.用法及び用量
通常、成人にはブリーバラセタムとして1日50mgを1日2回に分けて経口投与する。なお、症状により1日200mgを超えない範囲で適宜増減できる。
【静注製剤】
6.用法及び用量
ブリーバラセタムの経口投与から本剤に切り替える場合:
通常、ブリーバラセタム経口投与と同じ1 日用量及び投与回数にて、1回量を2分から15分かけて静脈内投与する。
ブリーバラセタムの経口投与に先立ち本剤を投与する場合:
通常、成人にはブリーバラセタムとして1日50mgを1日2回に分け、1回量を2分から15分かけて静脈内投与する。
いずれの場合においても、症状により適宜増減できるが、1日最高投与量は200mgとする。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書上、投与時期(タイミング)に関する情報はありません。
本剤は、食事の影響はほとんどありません。
参考情報
本剤の電子化された添付文書に以下の記載があります。
16.薬物動態
16.2 吸収
16.2.1 食事の影響
健康成人25例にブリーバラセタム50mgを空腹時又は高脂肪食の食後に単回経口投与したとき、空腹時と比べて、食後投与時では最高血漿中濃度到達時間(tmax)が約2.5時間延長し、最高血漿中濃度(Cmax)は37%低下したが、血漿中濃度-時間曲線下面積(AUC)は同等であった(外国人データ)。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
本剤は、単剤投与・併用投与 どちらも可能です。
参考情報
本剤の電子化された添付文書に以下の記載があります。
【経口製剤】
4.効能又は効果
てんかん患者の部分発作(二次性全般化発作を含む)
【静注製剤】
4.効能又は効果
一時的に経口投与ができない患者における、下記の治療に対するブリーバラセタム経口製剤の代替療法
てんかん患者の部分発作(二次性全般化発作を含む)
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤は、成人に対する用法及び用量のみ承認されています。(通常、成人の目安は15歳以上です。)
参考情報
本剤の電子化された添付文書に以下の記載があります。
9.特定の背景を有する患者に関する注意
9.7 小児等
小児等を対象とした臨床試験は実施していない。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に以下の記載があります。
16.薬物動態
16.4 代謝
ブリーバラセタムは主にアミダーゼにより加水分解されカルボン酸代謝物が生成し、副次的にCYP2C19により水酸化代謝物が生成する。また、カルボン酸代謝物の代謝にはCYP2C9が関与する。[10.参照]
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に、カルバマゼピンとフェニトインについて、以下の記載があります。
10.相互作用
10.2 併用注意(併用に注意すること)
■カルバマゼピン
臨床症状・措置方法:
本剤の血中濃度が低下するおそれがある。また、カルバマゼピンの活性代謝物であるカルバマゼピン-エポキシドの血中濃度が上昇し、副作用が増強されるおそれがある。
機序・危険因子:
カルバマゼピンが代謝酵素を誘導することにより本剤の代謝が促進される可能性がある。また、本剤がエポキシドヒドロラーゼを阻害することにより、カルバマゼピン-エポキシドの代謝が阻害される可能性がある。
■フェニトイン
臨床症状・措置方法:
本剤の血中濃度が低下するおそれがある。また、フェニトインの血中濃度が上昇し、副作用が増強されるおそれがある。
機序・危険因子:
フェニトインが代謝酵素を誘導することにより本剤の代謝が促進される可能性がある。また、本剤はフェニトインの代謝酵素を阻害する可能性がある。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書上、治療上有効な血中濃度に関する記載はありません。
参考情報
本剤のインタビューフォームに以下の記載があります。
Ⅶ.薬物動態に関する項目
1.血中濃度の推移
(1)治療上有効な血中濃度
該当資料なし
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
- ブリィビアクト静注25mg インタビューフォーム
本剤の電子化された添付文書上、薬物血中濃度モニタリング(TDM)の必要性に関する記載はありません。
参考情報
本剤のインタビューフォームに以下の記載があります。
I.概要に関する項目
2. 製品の治療学的特性
⑥ BRVの薬物動態プロファイルは用量比例性によって特徴付けられるため、治療用量のモニタリングは不要である。
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
本剤は、食事の影響はほとんどありません。
参考情報
本剤の電子化された添付文書に以下の記載があります。
16.薬物動態
16.2 吸収
16.2.1 食事の影響
健康成人25例にブリーバラセタム50mgを空腹時又は高脂肪食の食後に単回経口投与したとき、空腹時と比べて、食後投与時では最高血漿中濃度到達時間(tmax)が約2.5時間延長し、最高血漿中濃度(Cmax)は37%低下したが、血漿中濃度-時間曲線下面積(AUC)は同等であった(外国人データ)。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
本剤は、経口投与後速やかに血漿中濃度は最高値に達し、成人では投与2日目には定常状態に達すると推測されます。
参考情報
本剤のインタビューフォームに以下の記載があります。
Ⅶ.薬物動態に関する項目
1.血中濃度の推移 (2)臨床試験で確認された血中濃度
①単回経口投与(N01209試験)
日本人健康成人男性被験者(各群8例)にブリーバラセタム(BRV)2.5、10、25、50及び100mgを空腹時に単回経口投与したとき、すべての用量でBRV血漿中濃度は投与後ほぼ0.5~1.5時間に最高値を示し、消失半減期(t1/2)は投与量にかかわらず8~9時間あった。最高血漿中濃度(Cmax)及び血漿中濃度-時間曲線下面積(AUC)は用量比例的に増加した。
②反復経口投与(N01209試験)
日本人健康成人男性被験者(各群8例)にBRV5、20及び100 mg/日を単回(1~2日目)及び10日間(3~12日)反復経口投与したとき、血漿中濃度は共に投与後約0.5時間にCmaxに達した後、減少し、t1/2は約9時間であった。また、血漿中濃度は投与2日目には定常状態に達すると推測された。Cmax及びAUCは用量比例的に増加した。
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
本剤の電子化された添付文書に以下の記載があります。
18.薬効薬理
18.1 作用機序
ブリーバラセタムは、脳内のシナプス小胞蛋白質2A(SV2A)に高い親和性を示し、選択的に結合する。ブリーバラセタムとSV2Aの結合が発作抑制作用に寄与しているものと考えられている。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書、特定の背景を有する患者に関する注意の項に、腎機能障害患者の規定はありません。特別な注意喚起は行っておりません。
透析を実施中の患者を対象とした臨床試験は実施していないため、透析を実施中の終末期腎機能障害を有する患者でのデータはなく、ブリーバラセタムの使用は推奨されません。
参考情報
本剤の電子化された添付文書に以下の記載があります。
16.薬物動態
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
重度の腎機能障害を有する成人被験者に、ブリーバラセタム200mg(各群9例)を単回経口投与したとき、ブリーバラセタムの血漿中濃度-時間曲線下面積(AUC)は腎機能正常者と比較して21%上昇し、カルボン酸、ヒドロキシ及びヒドロキシ酸代謝物のAUCはそれぞれ3倍、4倍及び21倍に上昇した。これらの非活性代謝物の腎クリアランスは約1/10に低下した(外国人データ)
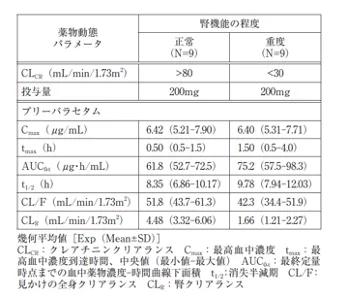
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書、用法及び用量に関連する注意の項に、用量調整に関する規定はありませんが、特定の背景を有する患者に関する注意の項に以下の記載があります。
肝機能障害患者へ投与する際は開始用量など適宜注意してください。
9.特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 肝機能障害のある患者(Child-Pugh分類A、B及びC)
本剤の血中濃度が上昇することがある。
参考情報
16.薬物動態
16.6 特定の背景を有する患者
16.6.2 肝機能障害患者
肝機能障害(Child-Pugh分類A、B及びC)を有する患者を対象にブリーバラセタム100mg(各群6 ~ 7例)を単回経口投与したときの薬物動態パラメータは以下のとおりであり、ブリーバラセタムの血漿中濃度-時間曲線下面積(AUC)は肝機能障害のない被験者と比較して、それぞれ50、57及び59%増加した(外国人データ)。[9.3.1 参照]
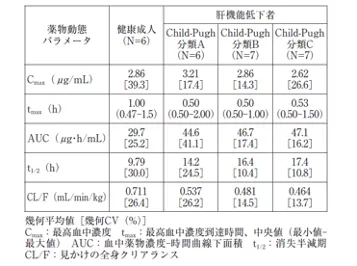
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に以下の記載があります。
9.特定の背景を有する患者に関する注意
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
ウサギを用いた胚・胎児発生毒性試験の240mg/kg(本剤の臨床最高用量200mg/日投与時の曝露量と比較して約6.7倍の曝露量)において、胚損失の増加、胎児体重の減少(対照群に対し6%)、矮小胎児の増加及び前肢あるいは後肢の骨端又は指骨の骨形成の不全又は欠損が認められた。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に以下の記載があります。
9.特定の背景を有する患者に関する注意
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。
ヒト乳汁中に移行するとの報告がある。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書、特定の背景を有する患者に関する注意の項に、高齢者の規定はありません。
特別な注意喚起は行っておりません。
参考情報
本剤の電子化された添付文書に以下の記載があります。
16.薬物動態
16.6 特定の背景を有する患者
16.6.3 高齢者
高齢者(65~79歳;クレアチニンクリアランス53~98mL/min/1.73m2)にブリーバラセタム400mg/日注)を1日2回10日間反復投与したときでは、65~75歳(10例)及び75歳超( 5 例)の消失半減期(t1/2)はそれぞれ7.9時間及び9.3時間であった。ブリーバラセタムの定常状態の見かけの全身クリアランス(CL/F)は若年健康成人男性(0.83mL/min/kg)よりわずかに低かった(0.76mL/min/kg)(外国人データ)。
注) 承認された用量は1 日50~200mg(分2)である。
本剤のインタビューフォームに以下の記載があります。
Ⅴ .治療に関する項目
(5) 患者・病態別試験
①健康高齢者を対象とした臨床試験(N01118試験:外国人データ)
健康高齢被験者 16例(男性8例、女性8例)が登録され、ブリーバラセタム(BRV)の投与を受けた。すべての被験者が白人で、年齢は65~75歳が10例、75歳超が6例であった。
副作用は14例(87.5%)に認められ、重症度はいずれも軽度であった。治験薬と関連のある死亡及び重篤副作用は認められなかった。また、その他の安全性評価項目において、臨床的に問題となる所見は認められなかった。
以上のことから、健康高齢被験者にBRVを投与した時の安全性及び忍容性は、若年者と同様であることが示された。薬物動態(PK)及び安全性の観点から、健康高齢男性及び女性被験者に対して BRVの用量調整は必要ないと考えられた。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
飲み忘れた場合は、気がついた時に1回分を飲んでください。ただし、次の飲む時間が6時間以内の場合は1回とばして、次の時間に1回分を飲んでください。絶対に2回分を一度に飲んではいけません。
引用
- ブリィビアクト錠25mg・錠50mg くすりのしおり
本剤の電子化された添付文書に以下の記載があります。
11.副作用 (一部抜粋)
11.2 その他の副作用
傾眠(14.9%)、浮動性めまい(10.9%)
【経口製剤】
17.臨床成績 (一部抜粋)
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第Ⅲ相試験(併用療法)
(抜粋)副作用発現頻度は、ブリーバラセタム50mg/日群で26.5%(40/151例)、200mg/日群で39.9%(59/148例)であった。主な副作用は、50mg/日群で傾眠9.3%(14/151例)及び浮動性めまい8.6%(13/151例)、200mg/日群で傾眠18.2%(27/148例)及び浮動性めまい10.8%(16/148例)であった。
17.1.2 国際共同長期継続投与試験
(抜粋)副作用発現頻度は、29.0%(60/207例)であった。主な副作用は、傾眠9.2%(19/207例)、浮動性めまい4.8%(10/207例)及び易刺激性2.4%( 5 /207例)であった。
【静注製剤】
17.臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第Ⅲ相試験(経口剤から注射剤への切り替え試験)
(抜粋)副作用発現頻度は60.0%( 6 /10例)であった。主な副作用は頭痛、傾眠が各20.0%( 2 /10例)であり、投与部位の副作用は血管穿刺部位紅斑10.0%( 1 /10例)であった。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤の電子化された添付文書に以下の記載があります。
13.過量投与
13.1 症状
外国の市販後に、ブリーバラセタムを1 回1400mg服用したときに傾眠及び浮動性めまいが発現したことが報告されている。
13.2 処置
本剤過量投与に対する特異的な処置薬はない。また、本剤の尿中排泄は10%未満であるため、血液透析は有効ではない。
引用
- ブリィビアクト錠25mg・錠50mg 電子化された添付文書
- ブリィビアクト静注25mg 電子化された添付文書
本剤を粉砕しての使用は適応外の用法となります。
詳細はインタビューフォーム XⅢ.備考をご確認ください。
本剤を粉砕した状態での薬物動態、有効性、安全性試験等は実施しておらず、それらを評価したデータはありません。
本剤の粉砕を推奨するものではありません。
XIII.備考 1. 調剤・服薬支援に際して臨床判断を行うにあたっての参考情報
(1) 粉砕
■ブリーバラセタム錠の粉砕後安定性試験
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム
簡易懸濁法を用いた本剤の経管投与は適応外の用法となります。
詳細はインタビューフォーム XⅢ.備考をご確認ください。
本剤を懸濁した状態での薬物動態、有効性、安全性試験等は実施しておらず、それらを評価したデータはありません。
本剤の懸濁投与を推奨するものではありません。
XIII.備考 1. 調剤・服薬支援に際して臨床判断を行うにあたっての参考情報
(2) 崩壊・懸濁性及び経管投与チューブの通過性
■ブリーバラセタム錠の簡易懸濁試験
引用
- ブリィビアクト錠25mg・錠50mg インタビューフォーム